Prion - Clinical syndromes, Kuru, CJD, vCJD, GBS, Fatal, Sporadic Familial Insomnia
Clinical syndromes/manifestation of Prion
Clinical syndromes/manifestation of Prion includes characteristics of TSE (Transmissible Spongiform Encephalopathy)
The incubation period is several years i.e. long the course of illness can last for months to years. TSE is a progressively debilitating neurological syndrome that is invariably fatal and is associated with pathological changes typically restricted to CNS.
There is an absence of specific immunological responses in the host. The syndromes from prions differ from virus encephalitis. The encephalopathy caused by prions denotes a pathologic process with no tissue inflammation and no immune response. Different prions affect different regions of the brain.
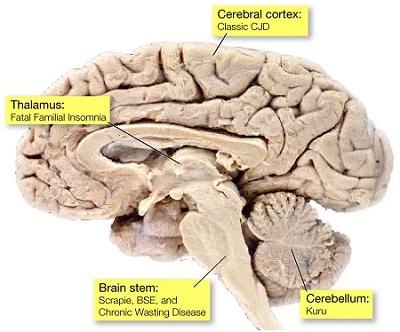
Cerebral cortex: symptoms include loss of memory and mental activity, also visual impairment (CJD)
Thalamus: fatal familial insomnia (FFI)
Cerebellum: lose control of body movements and difficulty walking (Kuru, GSS)
Brain stem: In mad cow disease (BSE), the brain stem is affected
Kuru
Kuru is a fatal neurological disease that is spread by cannibalistic funeral practice (the brain of a dead person consumed by a close female relative and children). It is caused by prions found in contaminated human brain tissue and the name Kuru means “to shiver” or “trembling in fear.”
The condition clinically is manifested as difficulty in walking followed by cerebellar tremors. Eventually, the tremor worsens, followed by progressive cerebellar ataxia and eventual death within a year of the onset of symptoms. Most die from pneumonia.
The clinical course lasts from 3 months to 2 years. The symptoms of Kuru include:
difficulty walking
poor coordination
difficulty swallowing
moodiness and behavioral changes
dementia
muscle twitching and tremors
inability to grasp objects, etc.
kuru occurs in three stages
1st stage:
exhibit some loss of bodily control
may have difficulty balancing and maintaining posture
2nd stage
inability to grasp objects
a person is unable to walk
body tremors and significant involuntary jerks and movements begin to occur
poor coordination
3rd stage
person in bedridden and incontienent
loss of ability to speak
dementia and behavior changes
starvation and malnutrition due to difficulty in eating and swallowing
Kuru is caused by Prion
Creutz-Jakob Disease (CJD)
CJD is the most common prion disease (85%). It is a subacute progressive encephalopathy characterized by rapidly progressive dementia associated with myoclonic jerks, memory loss, behavioral changes, and confusion. Symptoms include:
rapidly progressive dementia, leading to memory loss, personality changes, and hallucinations
anxiety
depression
ataxia (balance and coordination dysfunction)
aphasia (trouble speaking, reading, writing, understanding)
visual loss
hemiparesis (weakness of one entire side of the body)
speech impairment
jerky movements (myoclonus)
rigid postures and seizures
These symptoms are followed by involuntary movement. In terminal stages, the patient becomes mute and comatose. This condition is associated with extensive cortical spongiosis, gliosis, and neural loss. Death occurs in about 8 months, often of pneumonia.
CJD may also transmit from cuts in the skin. Types of CJD:
Familial (fCJD)
Sporadic (sCJD) – occurrence of disease in absence of infectious diseases or is hereditary
Variant (vCJD)
Latrogenic (iCJD)
iCJD arises as a result of medical procedures such as blood transfusion from an infected person, the use of human-derived pituitary growth hormones, gonadotropin hormone therapy, and corneal transplants.
The duration of these diseases greatly varies but non-inherited sCJD can be fatal within months (6 months)
Variant CJD (vCJD)
vCJD, caused by Prion, is a new disease infecting mostly adults aged 45. It is caused by the ingestion of BSE-infected beef products contaminated by neural tissues. This is because neural tissues have a much higher concentration of PrPC compared to other non-neural tissue.
Gerstmann-straussler-scheinker syndrome (GBS)
Gerstmann-straussler-scheinker, cuased by Prion, syndrome was first described in 1936. Clinical symptoms include slowly progressive limb, truncal ataxia, and dementia. Death occurs after 3-8 years of presentation of symptoms
Fatal familial insomnia
Fatal familial insomnia, caused by Prion, a clinical presentation varies from other syndromes but commonly includes- intractable insomnia, dysautonomia, dementia, and motor paralysis.vAs clinical presentation is varied, diagnosis is made by genotyping. Death occurs within 6 months-3 years following the clinical presentation.
.jpg)