Prion - Introduction, Dual Structure, Characteristics
Introduction to Prion
Prions are the shortened term for Proteinaeous infectious particles. They are small infectious proteins that cause fatal neurodegeneration in humans and animals and have no detectable nucleic acid. It causes various slow infectious diseases. The term ‘slow’ refers to the disease process and not the replication of the viruses.
Prions are unconventional agents, not viruses. Initially, prions used to be called slow viruses, infectious proteins, crystal proteins, infectious amyloidosis, and crystal proteins.
Some slow disease caused by prions includes:
Kuru
CJD (creutzfeldt Jakob Disease)
Variant CJD
Gerstmann-Straussler-Scheinker (GSS) syndrome
Fatal familial insomnia (FFI)
Sporadic fatal insomnia (f)
Scrapie (sheep)
Bovine Spongiform encephalopathy (BSE) = mad cow disease
Feline Spongiform encephalopathy
Transmissible mink encephalopathy
Chronic wasting of deer, mule, and elk
(1 to 6 = in humans; 7 to 11 = in animals)
Characteristics of Prion
Diseases caused by prions are a large group of related neurodegenerative conditions collectively known as TSE (Transmissible spongiform encephalopathies) and are associated with the accumulation of protease-resistant prion protein.
Misfolded/abnormally folded proteins in animals are pathogenic while normally folded versions are non-pathogenic. Prions cause similar proteins to also misfold. Some characteristics of prions include:
are filterable
lack of virion structure or genome
resistant to inactivation by heat, disinfectant, and radiation
do not elicit any specific host immune response
do not contain any nucleic acid
contains hydrophobic glycoprotein
difficult to decompose biologically so they survive in the soil for many years
they are inactivated by phenyl, ether, NaOH, and hypochlorite
In humans and animals, prions are called cellular prion proteins. It is similar to scrapie prion in its protein sequence but differs from it being protease sensitive and being present in the plasma membrane (scrapie prions are present in cytoplasmic vesicles).
The mutated and infectious form is built from the same amino acids but takes a different shape. The prion protein (PrP) gene found on the short arm of chromosome 20 of humans encodes a protein of 253a.a. termed PrPC (Prion protein cellular) is 100 times smaller than the smallest known virus.
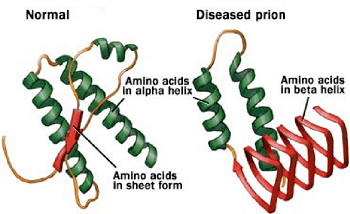
Fig: Two forms of Prions (Source: Wikipedia)
Two forms of prions
Both PrPC and PrPC are made up of exact same amino acid sequence but differ in their configuration.
PrPC is produced by normal healthy cells. It is also called “PrP-sen.” “sen” stands for protease sensitive. PrPC is mainly present in neurons in the brain but is also found in other cell types. The function of PrPC is believed to be involved in communication between neurons and controlling sleep patterns.
The 2nd form of prion (disease-causing form) is called PrPC. It is also known as “PrP-res”. “res” stands for resistant i.e. protease resistant. Amyloid fibers are toxic to cells and ultimately kill them (mostly brain tissues). Cells called astrocytes crawl through the brain digesting the dead neurons, and leaving holes where neurons used to be.